FDA User Fees: How Do They Work?
Nearly 30 years ago, the first law authorizing the Food and Drug Administration (FDA) to collect fees from drug manufacturers to expedite the review of new prescription drug applications was enacted. Since then, additional user fee programs for other human medical products (medical devices, generic drugs, biosimilars) and animal medical products (brand and generic veterinary drugs) have been authorized. User fees for new categories of products, from over-the-counter drugs to, most recently, e-cigarettes, are also regularly advocated for on Capitol Hill, with legislation to create new user fee programs often enjoying bipartisan support. This post will explore the genesis of FDA’s user fee programs and explain how they work, why they need to be reauthorized, and how businesses and patients alike can participate in the upcoming reauthorization process.
Congress has reauthorized the user fee programs every 5 years since their enactment and will be charged with doing so again in 2022 for all of FDA’s human medical product programs. Failure to reauthorize the programs to continue to operate would mean a catastrophic drop in funding for FDA, which would cause—in large part because more than 80% of the agency’s budget pays for personnel-related costs—massive layoffs of career staff who review new drugs and devices before they can be legally marketed and who monitor such products’ safety once on the market. The ultimate effect would be significant increases in the time it takes for new products to reach patients and health care providers, resulting in other undesirable downstream effects.
Although the existing user fee programs for human medical products are authorized through September 30, 2022, conversations will begin this year about what elements of the programs are working, what should be expanded, and what should be dropped. Those conversations will also determine the fees for various premarket submissions made to FDA (e.g., NDAs, BLAs, 510(k)s, PMAs, De Novos), how much funding the drug and device industries are willing to contribute, and, to a lesser extent, what Congress (i.e., taxpayers) will contribute.
Brief History & Mechanics of FDA User Fee Programs
FDA’s user fee programs are “pay-for-performance” programs. That’s because the agency’s authorizing statute, the Federal Food, Drug, and Cosmetic Act (FD&C Act), specifies certain timeframes for FDA to take action on a submission (e.g., 90 days to make a decision on a 510(k)), but the statute does not specify how often FDA must meet that mark. To many, the obvious interpretation is that FDA must adhere to the statute 100% of the time: when the law says 90 days, it means 90 days. What other law allows for wiggle room around such an explicit mandate?
Introducing performance into the mix allows for agency wiggle room in exchange for predictability for regulated industry. The construct changed from “the statute says FDA must make a decision in X days” to “FDA will make a decision on Y% of submissions in X days.” This was because in the pre-user fee era FDA was making decisions on very few submissions within the statutory timeframe: pre-user fee performance for some submissions was less than 50% (meaning FDA was reviewing less than half of the submissions it received in the time specified by statute). Because it was taking FDA so long to conduct reviews, drug and device manufacturers had little predictability to plan for marketing their products or conducting additional studies. This was bad for industry and worse for patients who sought to benefit from new drugs and devices that were being developed at a rapid pace, and particularly at a time when the public sought a much more robust government response to the beginning of the AIDS epidemic.
Despite discussions in the decades preceding enactment of the first law to set up some sort of fee program at FDA, industry and FDA in the early 1990s finally reached agreement on a plan for fees in exchange for commitments from FDA to render its decisions on new drug applications in a more timely and predictable fashion. The agreement required Congressional authorization and was enacted in 1992 as the Prescription Drug User Fee Act, known commonly as PDUFA (pronounced /p/-DOO-fuh). (User fee programs for medical devices, generic drugs, and biosimilars would follow in later years.)
PDUFA was an experiment; therefore, building in a sunset provision seemed wise. If the user fee program failed, Congress could let it expire without further action. The program proved successful, though, in part because of the sunset feature, which gave FDA, industry, patient groups, and Congress a chance to revisit the goals and commitments made on a periodic basis and to discuss what and how to improve.
The reauthorization process used today is prescribed by the FD&C Act. While the process for negotiating and reauthorizing each user fee program is identical, the steps described in the rest of this post are taken for each program individually, not collectively.
Overview of a Typical User Fee Reauthorization Process
The official reauthorization process begins with an initial public meeting at which the public is given an opportunity to express their views on the user fee program. Unofficially, trade associations, patient advocacy groups, and interested individuals spend the months leading up to the initial public meeting developing and refining ideas to present at the public meeting and at other times throughout the reauthorization process. The approximate date of this initial public meeting for the upcoming cycle can be predicted based on prior reauthorization schedules and is likely going to be held in Spring 2020.
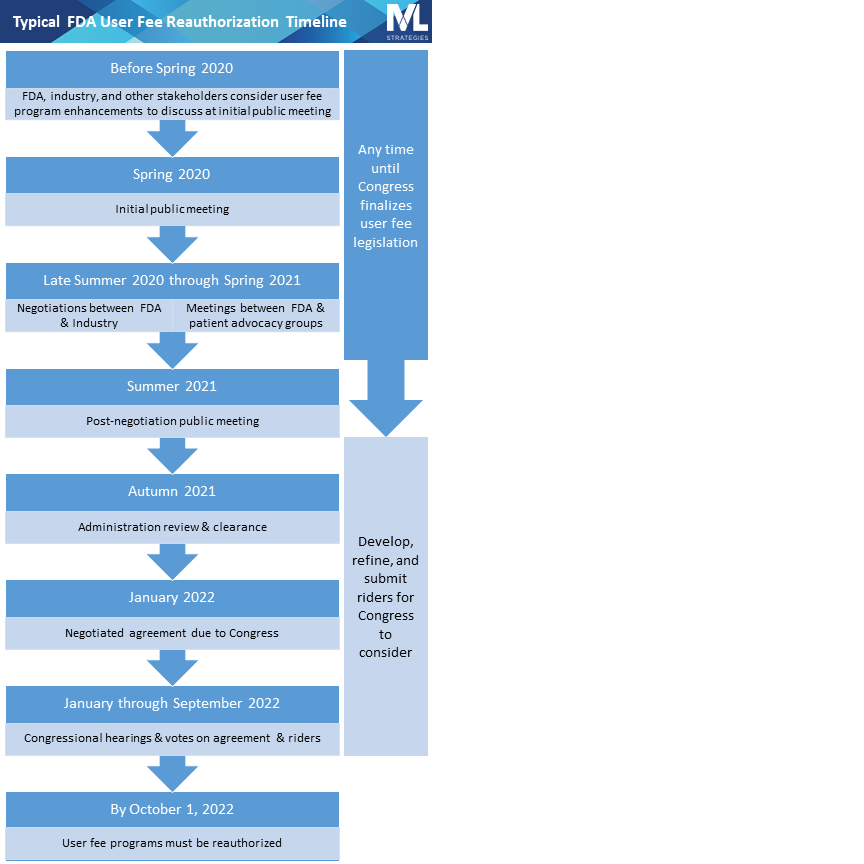
The initial public meeting is followed by negotiations between FDA and the regulated industry, typically represented by national trade associations. The drug industry negotiates PDUFA, the generic drug user fee program (called GDUFA), and the biosimilar user fee program (called BsUFA) primarily with representatives from FDA’s Center for Drug Evaluation and Research. The device industry negotiates the medical device user fee program (MDUFA) primarily with representatives from the Center for Devices and Radiological Health. The Center for Biologics Evaluation and Research, which has jurisdiction over some biologics and a few devices, participates in negotiations for PDUFA, GDUFA, BsUFA, and MDUFA. (Note that the acronyms for each law—PDUFA, MDUFA, etc.—are used colloquially to refer to both the law as well as the program.)
Negotiation meetings are complex discussions during which industry and the government each present their vision for the future of the user fee program and work together to balance multiple considerations related to fees, review timeframes, resource allocation, and other commitments. They are closed to the public and the press, though FDA publishes meeting minutes. During the same period in which FDA is negotiating with the industry, the agency will also meet with patient and consumer advocacy groups to hear their perspectives on the user fee programs.
The final output of the negotiation sessions for each FDA user fee program consists of:
- Recommendations for Congress to amend the law specifying the fee amounts for the next 5 years;
- The Commitment Letter, the common name for the document outlining review goals and other programmatic changes agreed to with the program’s other stakeholders; and
- Any other recommendations to Congress to change the law to effectuate certain programmatic elements agreed to in the negotiations.
Importantly, FDA cannot commit to meeting goals or changing programs in exchange for fees without Congressional ratification. The fees, irrespective of how eager industry is to pay them in exchange for FDA’s commitments to meeting the negotiated goals, need to be authorized and appropriated by Congress before the FDA can legally collect and spend them.
Following each negotiation is another public meeting convened by FDA at which the negotiated agreement is discussed and public feedback is received and considered. Then the agreement is sent to Congress, where it is analyzed and scrutinized as it moves through the typical legislative process (committees, votes, conferencing between House and Senate, passage) and then, finally, to the president for signature into law.
Why Does This Matter?
The reauthorization process offers opportunities for patients, consumers, manufacturers, health care providers, and many others to participate. The most effective way to have an impact is to have a compelling solution to an industry-wide or sector-wide problem that can be sold to a coalition of industry or patient representatives, to the FDA, or to Congress. Further, participating in the public meetings is a guaranteed way to share perspectives and ideas with other interested stakeholders. If an idea is not gaining traction with industry or the FDA, there is always the option of appealing directly to Congress.
Because the laws reauthorizing the user fee programs are so critical to the FDA’s funding, they are considered must-pass legislation. That means Members of Congress and other constituencies often pursue “riders,” which are legislative provisions that are not part of the agreement negotiated between FDA and industry but which are related to some aspect of FDA regulation and oversight of medical products (so-called because they “ride along” with the legislation reauthorizing the user fee programs).
Riders are a way for Congress to authorize or mandate that FDA implement a policy or change a program that is separate from the user fee agreement. Advocating for a legislative rider is a sure way to direct Congressional and agency attention to a public health or public policy issue even if it’s unrelated to the user fee agreement. Although the dream scenario is to get your rider enacted, increased attention to an issue often results even when the bill in question doesn’t get a ride on the user fee reauthorization law by virtue of a Congressional committee holding a hearing on the subject or other public activities related to advocating for it.
Our next post will briefly describe some of the changes made to the medical product user fee programs during the last reauthorization cycle, completed in 2017. We will also forecast what may be on the table as the current reauthorization cycle commences in earnest later this year.